Virtual Screening
An AI powered drug discovery and development solution for biopharma R&D to rapidly screen millions of molecules for a target protein and identify promising ones for downstream assays
Context
Evaluation process delays drug development
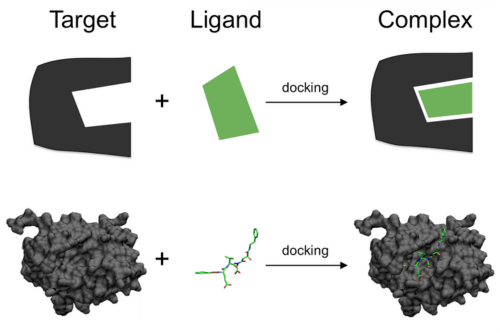
Virtual screening is an efficient tool used in early stages of drug development for the prioritization of compounds. Potential drug candidates need to be extensively screened for both their binding ability and functional properties.
At Aganitha, we have developed pipelines for Virtual Screening tailored for SMOLs and Antibodies. Continue to know more about the Small molecule virtual screening. To learn more about our solutions in Antibody virtual screening, click here.
Lack of high throughput infrastructure and scalability
Screening for promising candidates virtually has been around for decades. However, it is still not a major part of commercial pipelines due to lack of high throughput infrastructure and the inability to automatically scale up for high throughput computational requirements.
Inability to build custom solutions
Difficulty in building cross functional teams that understand diverse domains while having the relevant technology expertise which is necessary to develop custom solutions tailored for a specific purpose
Our Solution
An AI powered high throughput virtual screening solution aiding drug discovery and development
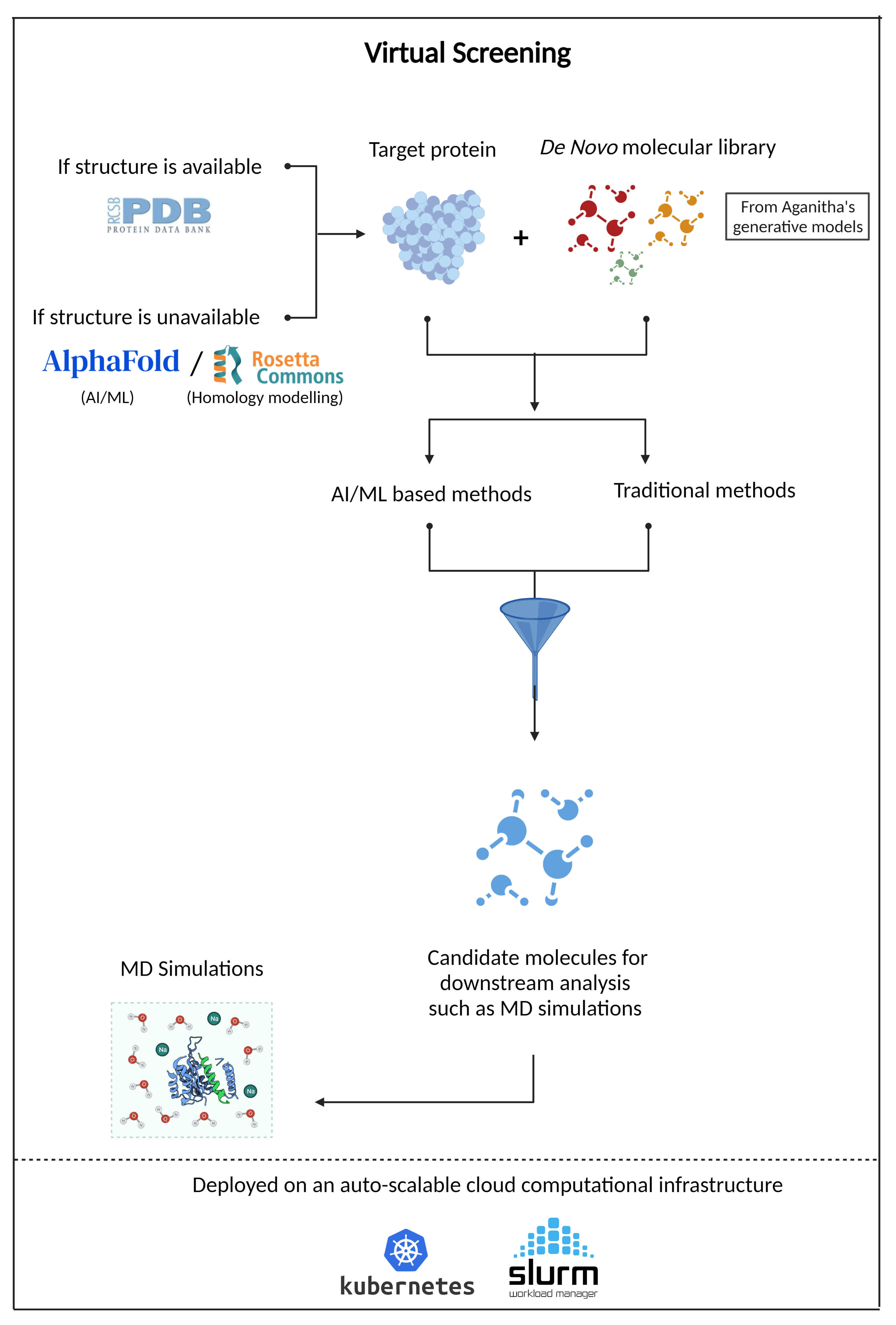
Aganitha’s solution combines the traditional molecular docking methods with modern AI/ML methods to predict the binding affinity
Molecular Docking Methods:
- Ligand preparation
- Protein preparation using the 3D structure of the protein. Homology modeling is used if the protein structure is not available. The solution also utilizes AlphaFold, a deep learning-based 3D protein structure prediction method.
Molecular Docking Methods Enhanced by AI/ML:
- Computational platform for high throughput screening and cloud-based Kubernetes environment with schedulers such as SLURM for auto-scaling and workload management.
- Modern AI/ML method with ECFPs (Extended Connectivity Fingerprints), and molecular graph convolutions to predict protein-ligand binding affinity – helping to screen millions of molecules for the target protein and shortlist appropriate molecules for downstream assays.
Highlights
Key components & strengths
Traditional molecular docking methods enhanced by AI/ML
ECFP, Molecular Graph Convolutions and Deep learning
High throughput Screening, Schedulers and Auto Scaling
HPC Cluster Environment and Custom Tools
Outcomes
Accelerated hit to lead optimization as part of the drug discovery and development process
Faster querying of structure activity relationships
Quick and Cost-effective Screening
Speeds up searching for potential drug molecule
Scaling of computing infrastructure
Discover our offerings across the biopharma value chain
Our Solutions
Our Services
Offering services in computational sciences and technology to complement biopharma R&D