Reaction Modeling
Context
Time consuming process and difficulty in estimating reaction yields is slowing down drug development
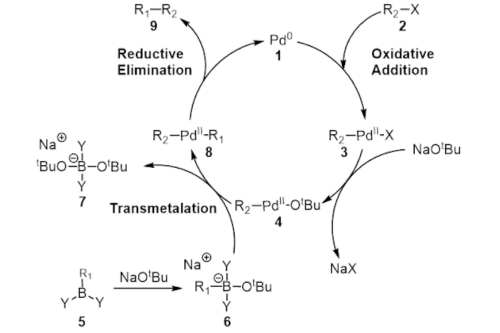
Yield optimization is one of the key priorities in the drug synthesis process development. Chemical and pharma companies have to perform various reactions in different conditions in a laboratory to find the optimal synthesis path. They further need to find the actual reaction yield (e.g., Suzuki Reaction) as it affects the evaluation of complex reaction paths and the selection of synthesis paths. One step with a low percent yield can lead to a big overall drop in yield.
Key challenges faced by the Chemical and Pharma companies when determining the reaction yield:
- Longer time to perform the reactions in a laboratory and determine the actual yield
- Lack of information about the yield of reactions that have not been reported previously in the literature
- Difficulties in optimizing synthesis paths from commercially available raw materials to form a significant ‘drug-like’ molecule
Our Solution
An AI powered drug discovery solution to predict reaction yields faster, and with higher accuracy
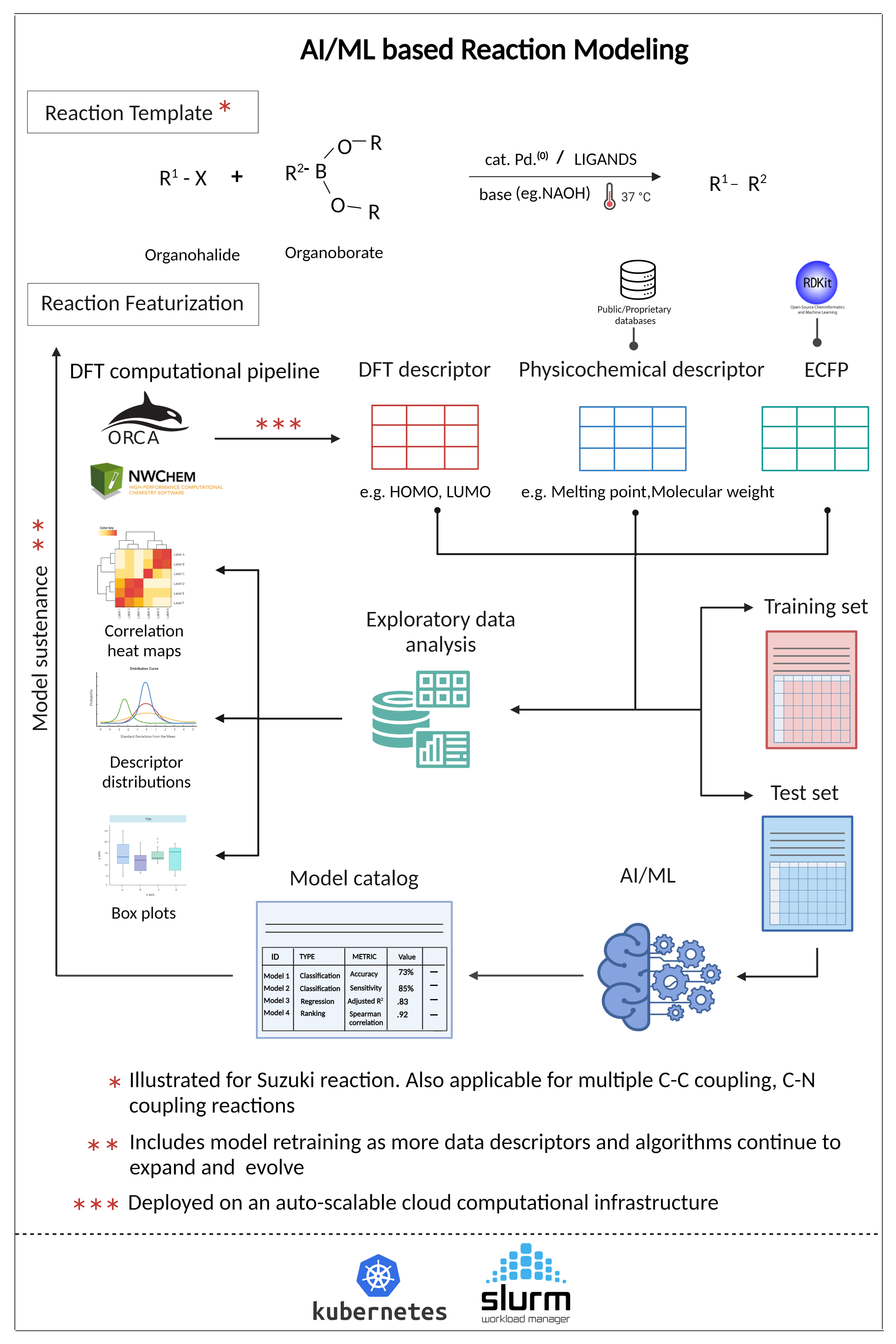
Aganitha’s ‘Reaction Modeling’ solution involves a combination of descriptors (Structural, Energy, ECFP, Reaction-mechanism based etc.) and model architectures (Random Forest, XG Boost, GCNs etc.) that can classify the reactions as high vs. low yield.
The solution features a data model that captures all the reaction information required for machine learning modeling. Further, its DFT computational pipelines speed up the estimation of reaction-mechanism based parameters such as energy, charge, bond-length, etc.
The Solution features:
- Scalable HPC pipelines for calculation of reaction-mechanism based DFT descriptors
- NLP techniques to extract information about reactants, products, solvents, catalysts, etc. from public datasets such as USPTO
- Data processing pipelines to filter reactions of classes e.g., Suzuki reaction coupling reactions
Highlights
Key components & strengths
Reactions Classifier
Reactions Explorer
Molecule Previewer
Model Catalog
DFT Pipeline
Scalable HPC Pipelines
Outcomes
Accelerated drug development through faster and accurate reaction yield prediction
Cost savings
Quick information retrieval
Visibility into expected yield
Faster and accurate
Discover our offerings across the biopharma value chain
Our Solutions
Our Services
Offering services in computational sciences and technology to complement biopharma R&D