SMOL Drug Design
AI-powered drug discovery platform for faster and cost-effective de novo small molecule design, characterization, optimization, and synthesis
Context
Time, effort, and cost intensive drug discovery process
The discovery of New Chemical Entities (NCE) with the desired biological activity is the foundational step for new therapeutics. This scientific exploration involves dealing with the twin challenges of an expanding druggable target protein space for which the vast chemical space (encompassing >1065 drug-like molecule) is to be investigated. Such challenges demand sophisticated algorithms to computationally aid R&D in a high-throughput scenario. For the biopharma industry, this is particularly relevant as it already faces a serious concern with Avoidable Experiment Expenditure (AEE), estimated at approximately $48 billion annually.
Advancements in cheminformatics, computational chemistry, and the integration of AI/ML are paving the way for an informed exploration of the chemical space and pushing the boundaries “beyond the rule of 5” (bRo5) chemical space. In silico techniques, driven by these advances, are proving to be significant cost-saving accelerators for failing fast before experimenting in the wet lab. De novo molecular generation enables optimization for range of properties such as ADMET, synthesizability, and not just biological activity. These methods help the discovery chemists to explore a vast chemical space in shorter time frames and enhance the quality of leads generated.
Our Solution
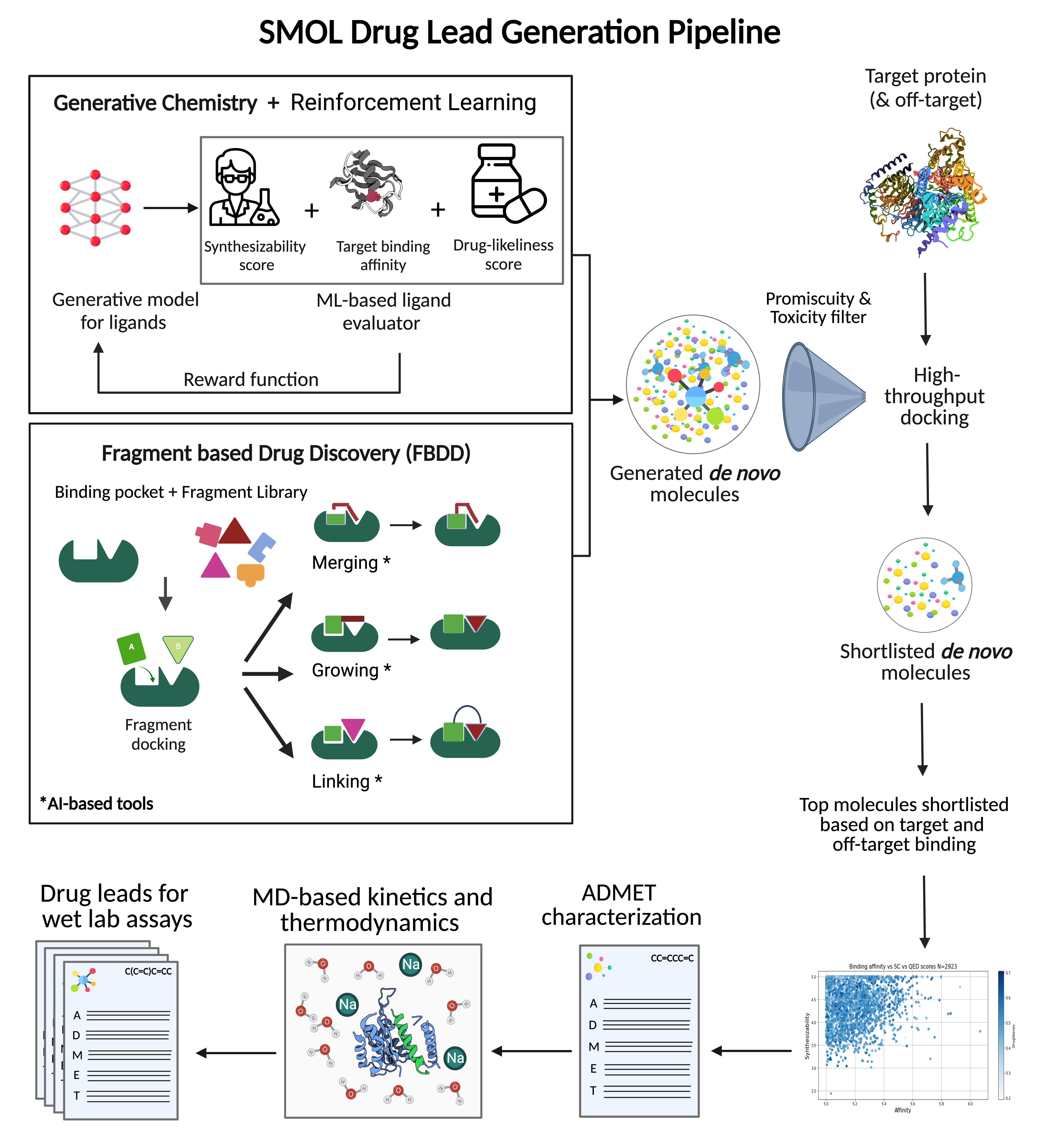
Highly repeatable and accelerated in silico pipeline to generate de novo molecular leads for a given target protein
We help reduce AEE for biopharma by combining the concepts of cheminformatics, computational and generative chemistry with advances in AI/ML, Cloud & DevOps. Our solutions complement the traditional lab-based approaches with in silico solutions to accelerate drug discovery. Key features of our solution include:
- Generative models to navigate the vast ‘drug-like’ chemical space while optimizing for biological activity, synthesizability, drug-like characteristics, ADMET properties
- Fragment Based Drug Design (FBDD) that leverages generative AI for fragment linking, fragment growing and fragment merging in the context of a binding pocket
- High throughput molecular docking to estimate binding affinity of lead candidates. Top docked complexes are further explored for potential interactions that reveal novel binding mechanisms through a curated binding site analysis
- Accelerated MD to study protein-ligand interactions and estimate binding strength
The workflows/pipelines can be customized for target proteins and molecular properties of interest. High throughput docking pipelines can be deployed on cloud-based Kubernetes environments with schedulers such as SLURM for auto-scaling and workload management.
Highlights
Key components & strengths
Virtual Screening
An AI-powered drug discovery and development solution for biopharma R&D to rapidly screen millions of molecules for a target protein and identify promising ones for downstream assays
Covalent Binders
Accelerate your covalent binder discovery for challenging protein targets with Aganitha
PROTACs
Targeted protein degradation by Proteolysis-Targeting Chimeras (PROTACs) for undruggable drug targets.
Reaction Modeling
Machine Learning powered computational chemistry methods to predict reaction yield
Crystal Structure Prediction
In silico discovery of polymorphs using Al/ML & GPU acceleration
Outcomes
Accelerate discovery of de novo SMOL drugs
Faster and Cost-effective
Low-effort Customization
Configurable and Scalable
Download our case study on using Molecular Dynamics in conjunction with biochemical assay data for hit-to-lead optimization
Discover our offerings across the biopharma value chain
Our Solutions
Our Services
Offering services in computational sciences and technology to complement biopharma R&D