Beyond the Hype: How AI and Computational Chemistry Are Reshaping Formulation and Manufacturing
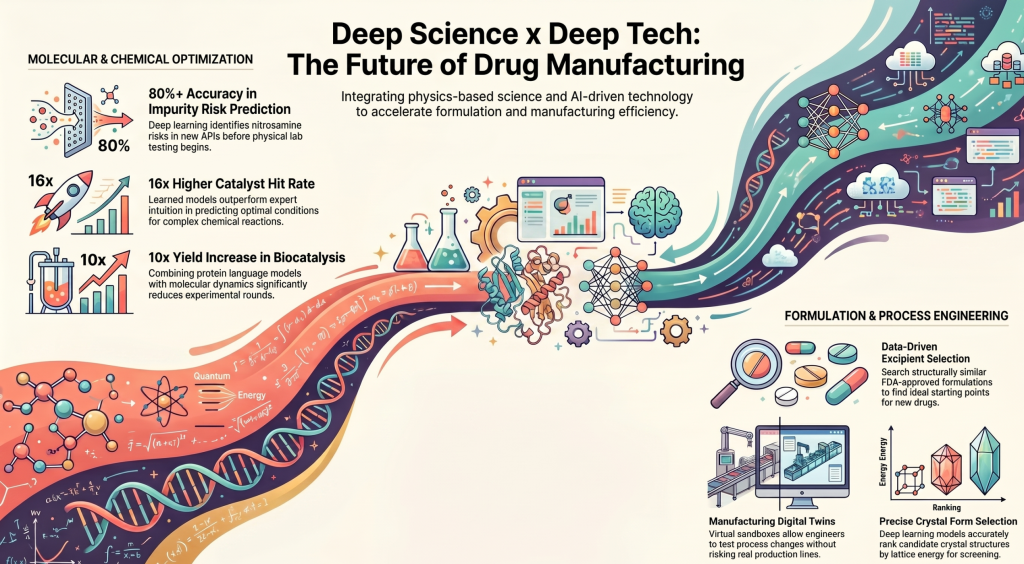
Where We Fit: Deep Science Meets Deep Tech
Our approach starts with a simple observation about the last twenty-five years of drug discovery. Two parallel revolutions have been unfolding side by side. On the “deep science” side, advances in genomics, structural biology, high-throughput experimentation, cheminformatics, and physics-based methods like quantum mechanics and molecular dynamics have steadily expanded what’s druggable, opening the door to newer modalities such as monoclonal antibodies, antibody-drug conjugates, PROTACs, engineered enzymes, and cell and gene therapies. On the “deep tech” side, generative, predictive, and increasingly agentic AI, paired with HPC, cloud computing, and GPU acceleration, has given scientists tools to navigate that expanding complexity at a pace that wasn’t possible a decade ago.
Our core thesis is that the real value comes from integrating these two threads. We apply that integration across disease biology research, de novo molecule design, drug candidate optimization, and chemistry, manufacturing, and controls (CMC) and process R&D.
Our work in Formulations and CMC spans four broad areas:
- predicting molecular properties such as toxicity, pKa, solvent behavior, and peptide permeability;
- modeling catalysis and biocatalysis, including yield prediction, enzyme engineering, and ligand optimization;
- supporting formulation design through tools like API surrogate search and crystal structure prediction; and
- modeling process and unit operations, from antibody titer forecasting to digital twins of manufacturing steps.
Molecular Property Modeling: Predicting Risk and Behavior Earlier
A recurring theme in this category is catching problems computationally before they show up in the lab. One example is our nitrosation propensity prediction tool, where a hybrid of knowledge-based rules and deep learning estimates the nitrosation risk of new APIs and excipients with better than 80% accuracy, a meaningful safeguard given how much regulatory attention nitrosamine impurities have drawn in recent years.
Our pKa estimation protocol applies the same rigor. We use density functional theory (DFT) derived energies calculated on solvent-specific conformer ensembles, which gives accurate pKa prediction across multiple industrial solvents. We’ve also packaged this capability as a standalone predictive model.
Our solubility prediction work takes a different computational route, using graph neural networks to rank candidate solvents for drug-like organic molecules, which helps narrow down solvent screening before committing bench time. And for the growing class of cyclic peptide therapeutics, we built CycPepPer, a tool that predicts membrane permeability using a directed message-passing neural network architecture, reaching an ROC-AUC of 0.88, a strong signal for a property that’s traditionally been difficult to model.
Catalysis and Biocatalysis: Fewer Experiments, Better Outcomes
For enzyme engineering, we combine protein language models with molecular dynamics simulations to guide variant selection, and we’ve achieved yield increases exceeding 10x while substantially cutting down the number of experimental rounds needed to get there. That’s a direct translation of computational insight into lab efficiency.
We see a similar pattern in catalyst and reagent selection. Our deep learning architecture, which models the interactions between reactants and reagents, delivers a 16x higher hit rate than human subject matter experts in predicting optimal conditions for cross-coupling reactions, showing that learned models can outperform expert intuition on condition selection for certain reaction classes. On the mechanistic side, we use DFT-based investigation of reaction pathways to understand selectivity and drive C–O coupling optimization, pairing data-driven prediction with first-principles explanation.
Formulation Design Support: Compressing the Search Space
Formulation work tends to be full of empirical trial-and-error, and several of our tools target that directly. Our API surrogate search gives formulation scientists a sensible starting point for excipient selection by searching for structurally and physico-chemically similar APIs across the universe of FDA-approved formulations, putting decades of precedent at their fingertips.
For amorphous solid dispersions, a major focus area given how many modern APIs are poorly soluble, we use molecular dynamics protocols to estimate glass transition and melting temperatures for APIs, polymers, and their binary mixtures, with results that correlate well against experimental trends. The same simulations let us probe molecular interactions inside a formulation, such as hydrogen bonding, radial distribution functions, and solvent-accessible surface area, across different drug loads, giving us a mechanistic view of why a particular polymer-API combination behaves the way it does.
Beyond ASDs, we’ve built a computational tool that predicts API true density, a property that matters directly for powder porosity and dissolution behavior. We address liquid-liquid phase separation, often called “oiling out,” through a thermodynamic approach that predicts the phase separation boundary during crystallization, giving formulation teams a model-driven path through crystallization design. And for crystal form selection, our deep learning models provide accurate rank ordering of candidate crystal structures by lattice energy, supporting decisions in polymorph and salt screening.
Process and Unit Operations Modeling: Looking Ahead in Manufacturing
On the manufacturing side, our mAb titer forecasting work uses historical production data to build models that predict final-day titers, giving operations teams warning that helps them manage disturbances before they cascade into downstream purification issues. More broadly, we build digital twins that combine mechanistic models with AI and machine learning to identify critical process parameters and model individual unit operations, giving process engineers a sandbox for testing changes before they touch the real production line.
The Throughline
What ties all of these examples together is how we build computation into pharma R&D: as purpose-built models grounded in the underlying physics and chemistry of each problem, whether that’s DFT for pKa, GNNs for solubility, or mechanistic digital twins for unit operations. The accuracy figures and hit-rate improvements above point to a consistent message: when we integrate deep science and deep tech deliberately, the payoff is measurable today, in fewer experiments, faster screening, and more predictable manufacturing outcomes.
Want to explore how these capabilities could apply to your formulation or manufacturing challenges? Get in touch with us: info@aganitha.ai